My main research interest is to understand the structural and thermodynamic properties of biomolecular systems. These include the inhibition of the activity of a protein, the complexation of toxic metal ions, just to name a few. To address this I use classical and quantum computational approaches.
The development of computational methods to calculate a priori physico-chemical properties of biomolecular systems is essential to support rational design and development in biotechnology.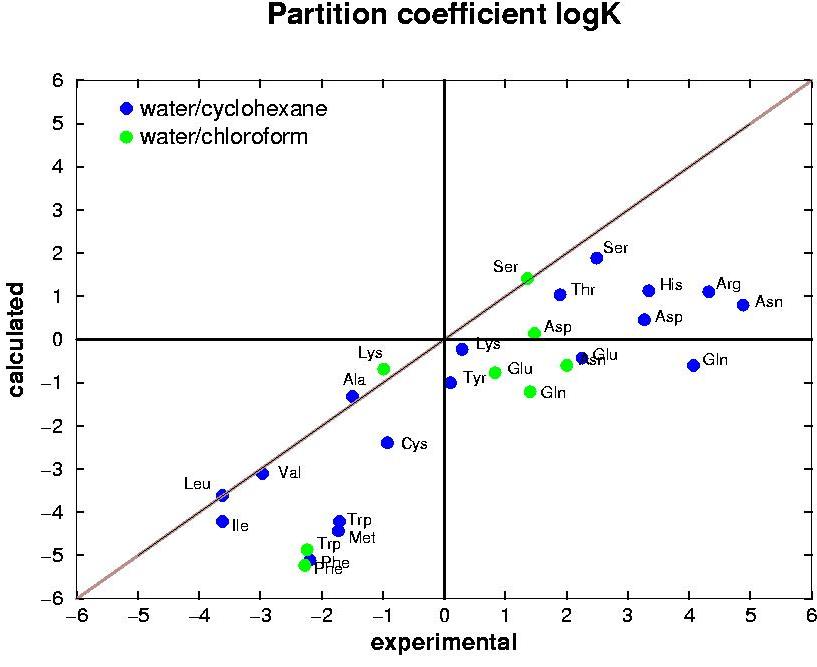 A critical
requirement of any molecular model or force field used for biomolecular
simulations is to correctly reproduce not only the conformational
properties of the molecules involved but also the solvation and
partitioning behaviour of specific functional groups in, and between,
different environments. The
partition
properties of amino acid analogues have
been
evaluated via free energy calculations using GROMOS96 force field
(43a2) (J.
Comput. Chem 2002, 23, 548).
A critical
requirement of any molecular model or force field used for biomolecular
simulations is to correctly reproduce not only the conformational
properties of the molecules involved but also the solvation and
partitioning behaviour of specific functional groups in, and between,
different environments. The
partition
properties of amino acid analogues have
been
evaluated via free energy calculations using GROMOS96 force field
(43a2) (J.
Comput. Chem 2002, 23, 548).Force field parametrization could also be based on the free entalpies of solvation (J. Comput. Chem, 2004, 25, 1656.)
GROMOS force field 53a5 and 53a6 in GROMACS format.
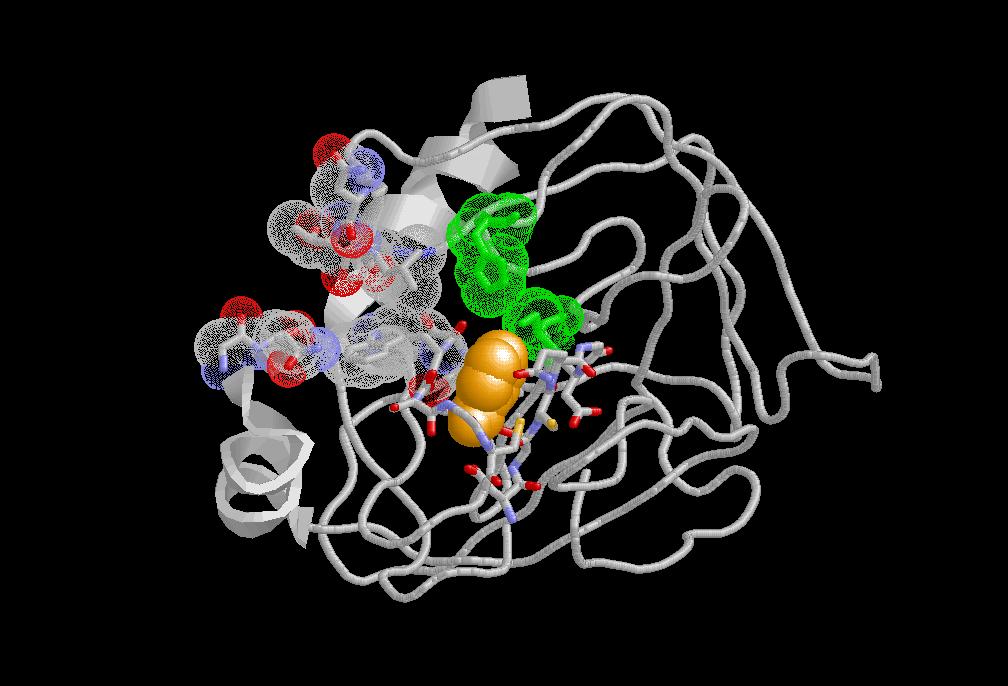 Advances in the
field of computational chemistry have paved the way
for calculating free energy differences associated with various chemical processes. Among others, these include the
recognition of a ligand by a receptor and the
solvation of a molecule. One common and
theoretically rigorous method to estimate the difference in the free energy between two states of a system is the coupling
parameter approach in conjunction with molecular
dynamics simulation techniques using either
the thermodynamic integration or
thermodynamic perturbation formula. Recurring
questions in such calculations are about
the treatment of the environment and about sampling
and convergence.
Advances in the
field of computational chemistry have paved the way
for calculating free energy differences associated with various chemical processes. Among others, these include the
recognition of a ligand by a receptor and the
solvation of a molecule. One common and
theoretically rigorous method to estimate the difference in the free energy between two states of a system is the coupling
parameter approach in conjunction with molecular
dynamics simulation techniques using either
the thermodynamic integration or
thermodynamic perturbation formula. Recurring
questions in such calculations are about
the treatment of the environment and about sampling
and convergence. The effect of the inclusion of explictly ions in the calculation was investigated. (J. Comput. Chem., 2005, 26, 115.) A large set of inhibitors to serine proteases was used to study the sampling and convergence in binding free-energy calculations (Journal of Computer-Aided Molecular Design, 2003, 17, 673.)
Computational
approaches such as molecular dynamics simulation techniques can be used
to understand inter and intra molecular interactions in peptide systems
in atomic details. This has high relevance when the complete
characterization of the systems is diffult to archieve using the
standards experimental techniques such as X-ray and liquid-state NMR.
A example
is the study of the polymorphism of amyloid peptide
PrP106-126. Other projects
regard the formation and stabity of
designed coiled coil peptide.
Lanthanide ions
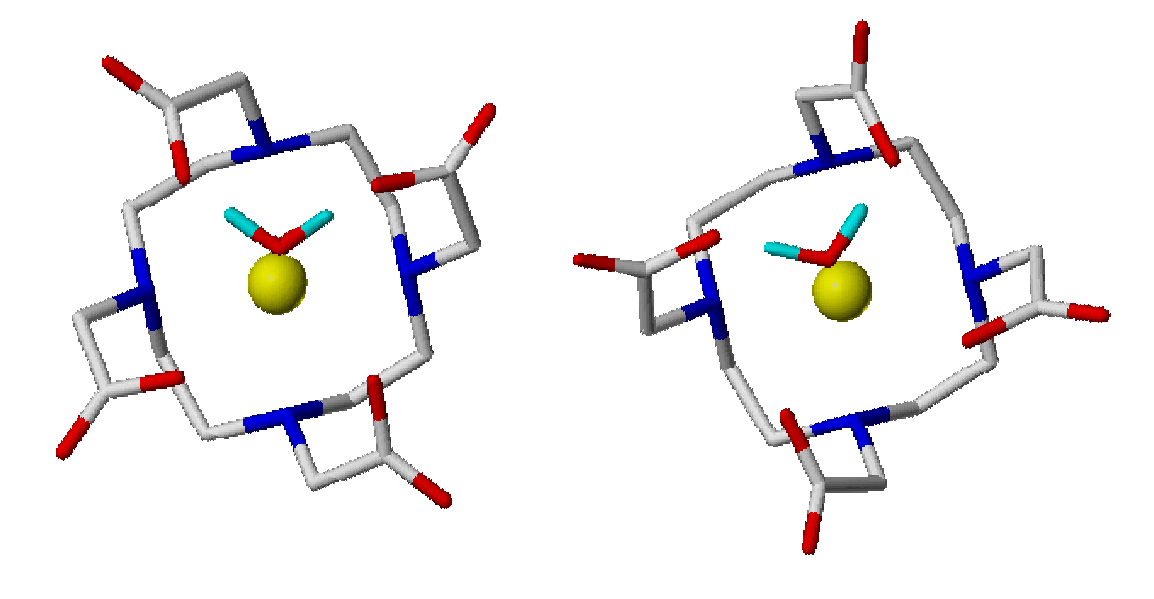 Lanthanide(III)
complexes, in
particular
Gadolinium complexes, are employed as contrast agents in Magnetic
Resonance Imaging diagnostic techniques. To
address the structural and dynamical properties of these compounds
classical and ab initio method
have been used.
Lanthanide(III)
complexes, in
particular
Gadolinium complexes, are employed as contrast agents in Magnetic
Resonance Imaging diagnostic techniques. To
address the structural and dynamical properties of these compounds
classical and ab initio method
have been used. The choice of the effective core potential (ECP) determines the reliability of the results and the inclusion of solvent effects was required for a correct description of several molecular properties (JACS 2002, 124, 4901)
The Gd-ligand interactions within a molecular-mechanics force-field have been parameterized starting from ab initio results (J. Phys. Chem. A. 2000, 104, 3421).