


"The Role of Chemistry Across Disciplines From Humanities to Life Sciences in Understanding Complexity and Emergence" in Angewandte Chemie International Edition
A group of researchers from the humanities, economics, social sciences, natural and life science developed a definition of the topics complexity and emergence that can be applied across disciplines. Here, concepts of complexity and emergence in chemistry and biochemistry are discussed, to promote a discourse between the natural and life sciences and philosophy. Although chemical research often employs reductionist strategies, the properties of molecules and their linked functions exhibit emergent properties that cannot be inferred solely from their atomic constituents. Assembly theory and the work of Manfred Eigen offer ways to quantify and predict emergence in chemistry, particularly in relation to the origins and evolution of life. This review emphasizes the chemical prerequisites for life, such as the formation of natural products, the emergence of nucleic acids that carry information, and the functional roles of proteins. From a philosophical standpoint, modern ontology provides a means of understanding reality that is both process-based and subject-independent. By integrating chemistry, biology and philosophy, the synopsis of this review addresses the predictive, post facto and historically unique aspects of complex systems, offering a conceptual framework for comprehending the emergence of molecular function and the evolution of living systems.

"Switching Shapes: Reversible Three Species Photoisomerization of Substituted 1,2-Dihydro-1,2-azaborinines" in Journal of the American Chemical Society
Derivatives of 1,2-dihydro-1,2-azaborinines generally undergo selective photochemical electrocyclic ring-closure reactions to the corresponding Dewar isomers (2-aza-3-borabicyclo[2.2.0]hex-5-ene). Depending on the substitution pattern, these photoreactions can also yield benzvalene (3-aza-4-boratricyclo[3.1.0.02.6]hexane) analogues. Here, we report the synthesis of 1,2,3,5-tetrasubstituted dihydroazaborinines by transition-metal-catalyzed late-stage functionalization and the investigation of their photophysical and photochemical properties using transient absorption spectroscopy. The introduction of aryl groups at the 3- and 5-positions induces a pronounced bathochromic shift of the absorption maximum. Under broad-spectrum irradiation (280–400 nm), quantitative conversion to the benzvalene isomer can be achieved. The initial photoisomerization proceeds via excitation to the short-lived singlet excited state (S1) yielding the Dewar isomer, whereas the subsequent conversion of this intermediate occurs through a long-lived excited state. Notably, the second isomerization step is accompanied by an interchange of the carbons C3 and C4. Once formed, the benzvalene isomers exhibit exceptional thermal stability. Cycloreversion to the Dewar isomer and even to the dihydroazaborinine structure can be triggered photochemically through targeted excitation and during both processes the substituents return to the C3 and C5 positions. The thermal cycloreversion of the benzvalene isomer can yield either the educt BN-benzene isomer (1,2,3,5-substitued) or its 1,2,4,5-substituted isomer. Computational studies revealed a stepwise mechanism for the thermal back reaction reforming the educt, while a concerted, energetically less-favorable pathway leads to the 1,2,4,5-substituted analogue.

"Dispersion-Controlled Excited-State Dynamics in Azobenzene Photoisomerization" in Journal of the American Chemical Society
Weak interactions, like London dispersion forces, are cumulative in nature and have been thought to be essential for only the structure and stability of large molecular systems. Only recently has their relevance for chemical reactivity been recognized. Until today, their role in photoreactions and subsequent ultrafast excited-state processes has remained elusive. Here, we show the impact of London dispersion on the excited-state behavior and the outcome of the photoreaction of the iconic photoswitch azobenzene as a representative example. Increased dispersion interactions between substituents decisively prolong the excited-state lifetimes by preventing direct passage through the conical intersection. This significantly alters the dynamics of the Z to E photoisomerization. We expect our findings to lead to increased research interest in such “dispersion-controlled excited-state dynamics” relevant for the steering of ultrafast processes.

"Trapped Together: How a Second Phase Induces Extrinsic Self-Trapping Leading to Enhanced Emitting States in 2D Perovskites" in Chemistry of Materials
Two-dimensional (2D) lead halide (R2PbX4, with X = Br, Cl, and R = organic ammonium cation) perovskites (PSK) are promising materials for the generation of white light due to self-trapping events leading to broadband photoluminescence (PL). In the mixed-halide series NEA2Pb(Br1−nCln)4 (NEA = 2-naphthyl 2-ethylammonium), we systematically investigated defect effects on optical properties. We focused on Cl-rich samples, which showed enhanced broad emission, and compared the chloride-only component with two mixtures, n = 0.7, which exhibits one phase, and n = 0.5, including a small quantity of an additional Br-rich phase. We combined X-ray diffraction with steady-state and time-resolved optical studies and found prolonged PL and recombination lifetimes for n = 0.5 > 0.7 > 1, demonstrating the enhancing effect of defect-induced extrinsic self-trapping on emitting states.

"Visible-Light Photoswitchable Covalent Tetra-Ortho-Fluoro-Azobenzene Carbon Nanodot Hybrids for Optostimulation" in ChemPhotoChem
Carbon nanodots (CNDs) have attracted growing interest due to their potential applications in sensing, imaging, and optically controlled bio-applications. Herein, the covalent functionalization of citric acid/ethylenediamine-based CNDs with a tetra-ortho-fluoro-azobenzene derivative (F-Azo) is presented. This approach aims to integrate the intrinsic photoluminescence of CNDs with the reversible photoisomerization properties of F-Azos triggered by visible light. The CND-F-Azo hybrids are synthesized via a terminal carboxylic acid group located on the F-Azo, which can be attached via amide coupling to surface-accessible amines on the CNDs. The structural and optical characterization of the resulting hybrid material is performed using a variety of analytical and spectroscopic techniques, as well as computational analyses supporting the covalent linking between the molecular and nanomaterial components and the interactions existing between them. In order to assess the impact of functionalization on physicochemical properties, the hybrid is further analyzed with respect to zeta potential, lipophilicity, and cell viability using HEK-293 cell assays. To assess cellular uptake and intracellular localization, confocal fluorescence imaging is employed. This work contributes to the development of light-responsive nanomaterials with tailored surface properties, highlighting the potential of Azo-functionalized CNDs as multifunctional platforms for future in vitro and in vivo optostimulation applications.

"Alternating Orthogonal Switching in a Thiophenyl-Phenyl-Bis-Azobenzene Switch" in Chemistry – A European Journal
Abstract To design efficient molecular information storage systems with multi-photoswitchable entities, orthogonal isomerization of the different switchable moieties is essential. Various challenges, like unintentional energy transfer, spectral overlap, and other energy dissipation channels, have to be addressed by intelligent molecule design. In this context, we took advantage of calculations to design a bis-azobenzene switch, which consists of a phenyl- and a thiophenylazobenzene moiety in meta-connection to reduce π-conjugation. Ultrafast spectroscopy and computational studies confirmed that this bis-photoswitch exhibits alternating orthogonal switching behavior when irradiated with light of different wavelengths. These results represent a significant advancement toward the development of efficient and adaptable organic multi-photoswitches for applications, such as information storage, molecular machines, or smart materials.

"Breaking Bonds with Short-Wave Infrared Light: BODIPY Photocages for Two-Photon Activation in the 900–1500 nm NIR-II Window" in Journal of the American Chemical Society
In this collaborative study, we investigated 11 BODIPY-based photocages for two-photon excitation within the first and second biological windows, aiming to overcome the tissue penetration limits of UV/visible activation. By modifying the 3- and 5-positions, we identified structural motifs, such as strong charge-transfer character and increased vibrational freedom, that significantly enhance two-photon absorption in the 900–1500 nm range. These photocages enable photorelease using two SWIR photons carrying as little as 20 kcal/mol each, establishing key design principles for NIR-II photoactivatable systems.
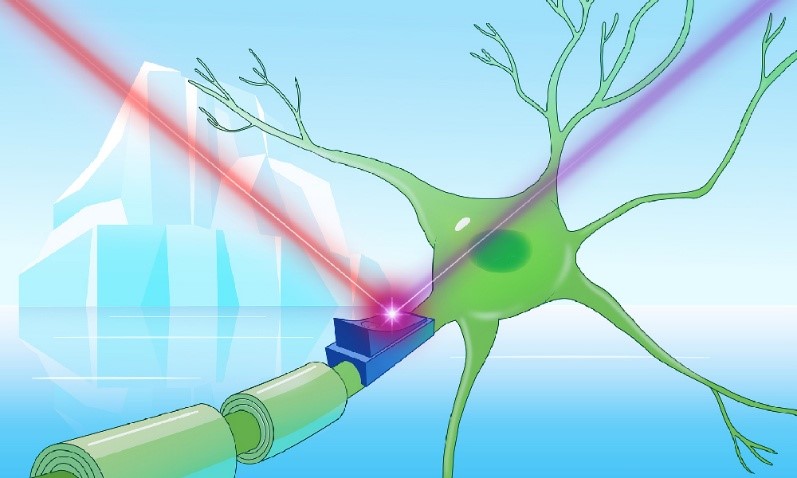
"CryoRhodopsins: A comprehensive characterization of a group of microbial rhodopsins from cold environments" in Science Advances
Comprehensive study of a new group of microbial rhodopsins called CryoRhodopsins (CryoRs), whose members mainly originate from cold environments. By combination of insights gaining from single-particle cryo-EM, X-ray crystallography, and time-resolved spectroscopy we elucidated, that the characteristic arginine residue of this group (R57 in CryoR1) stabilizes the UV-absorbing M2 intermediate state, which causes the extremely slow photocycle dynamics of CryoRs at neutral and alkaline conditions. Furthermore, the mechanism causing the photoswitch-like behavior was elucidated. All in all, the data suggests that CryoRs function as sensors for UV irradiation, as illustrated by the population of the UV sensitive M2 intermediate upon irradiation by sunlight.

"Covalent Carbon Nanodot-Azobenzene Hybrid Photoswitches: The Role of Meta/Para Connectivity and sp³ Spacer in Photophysical Properties" in J. Mater. Chem. C.
In this collaborative study, we investigated the properties of azobenzene-functionalized carbon nanodots, resulting in azobenzene-nanoparticle hybrids. Three distinct hybrids, differing in meta and para connectivity as well as linker length, were examined. The findings demonstrate that both the functionalization pattern and connectivity type significantly influence the efficiency of energy and charge transfer from the carbon dot to the azobenzene moiety, thereby affecting the overall optical properties. These hybrid systems show strong potential for diverse applications, including optostimulation and biomedical use.
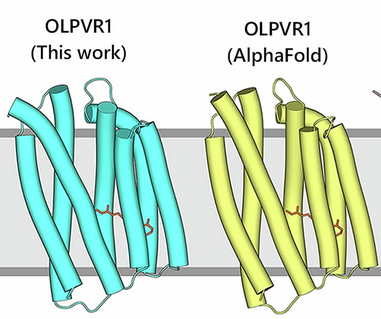
"Ion-conducting and gating molecular mechanisms of channelrhodopsin revealed by true-atomic-resolution structures of open and closed states" in Nat. Struct. Mol. Biol.

"Multistate Dihydroazulene-Spiropyran Dyads: Path-Dependent Switchings and Refinement of the “Meta-rule” of Photoactivity" in Chem. Eur. J.
Dihydroazulene-spiropyran (DHA-SP) photochromic dyads were prepared and investigated by stationary and ultrafast spectroscopies. The dyads can reach eight different states, some only by a specific sequence of stimuli. This path-dependent switching adds an additional degree of data storage.


"Energy transfer booster: how a leaving group controls the excited state pathway within a caging BASHY–BODIPY dyad" in Phys. Chem. Chem. Phys.